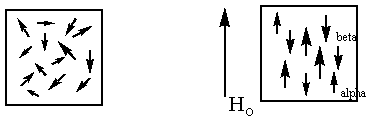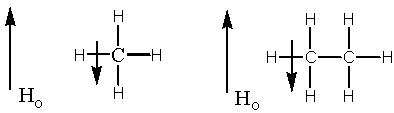|
|
random orientation |
alpha and beta orientation in field |
By Maryellen Nerz-Stormes
[Back to Study Aids]
Nuclei possessing angular moment (also called spin) have an associated magnetic moment. A few examples of magnetic isotopes are 13C, 1H, 19F,14N, 17O, 31P, and 33S. Please note that not every isotope is magnetic. In particular, you should note that 12C is not magnetic. If a nucleus is not magnetic, it can't be studied by nuclear magnetic resonance spectroscopy. For the purposes of this course, we will be most interested in 1H and 13C. I will limit my discussions to 1H in this short treatment. Generally speaking, you should think of these special nuclei as tiny, atomic, bar magnets.
Nuclear Magnetic Spectroscopy is based on the fact that when a population of magnetic nuclei is placed in an external magnetic field, the nuclei become aligned in a predictable and finite number of orientations. For 1H there are two orientations. In one orientation the protons are aligned with the external magnetic field (north pole of the nucleus aligned with the south pole of the magnet and south pole of the nucleus with the north pole of the magnet) and in the other where the nuclei are aligned against the field (north with north, south with south). The alignment with the field is also called the "alpha" orientation and the alignment against the field is called the "beta" orientation. From my description of the poles, which orientation do you think is the preferred or lower in energy? If you guessed the "alpha", you are correct. It might be worth noting at this point that before the nuclei are placed in the magnetic field they have random orientation
|
|
random orientation |
alpha and beta orientation in field |
Since the alpha orientation is preferred, more of the population of nuclei are aligned with the field than against the field. You might wonder why any spins would align against the field. Realize that we are talking about atomic magnets. These are very, very weak magnets. The energy difference between the alpha and beta orientations is not large. There is enough energy for nuclei to exchange between the two orientations at room temperature, though a slight excess on average is in the lower energy, alpha state.
The nuclear magnetic resonance (NMR) spectroscopy experiment involves using energy in the form of electromagnetic radiation to pump the excess alpha oriented nuclei into the beta state. When the energy is removed, the energized nuclei relax back to the alpha state. The fluctuation of the magnetic field associated with this relaxation process is called resonance and this resonance can be detected and converted into the peaks we see in an NMR spectrum.
What sort of electromagnetic radiation is appropriate for the low energy transition involved in NMR? Well believe it or not, radio waves do the trick. Radio waves are at the very low energy end of the electromagnetic spectrum and are sufficient to induce the desired transition. It is for this reason that NMR is considered to be a safe method of analysis. The same technology is now used in hospitals in MRI (Magnetic Resonance Imagining - people are afraid of the word nuclear). If you have ever had an MRI done, realize that you were placed in a magnetic field and all your magnetic nuclei lined up in the manner described above. Excess nuclei were pumped to higher energy states as you were exposed to radio waves.
The following are two very, very important points to accept and learn if you are going to understand the rest of the discussion.
1. Electric currents have associated magnetic fields.
2. Magnetic fields can generate electric currents.
If you haven't had physics yet, try to accept these two points. Certainly most people have at least heard of electromagnets and if so, you probably have some idea about the first statement.
The following is a very important NMR relationship. This expression relates the external field to the frequency of resonance.
| n= | mHo |
| 2p |
In this equation, n is frequency, m is the magnetogyric ratio (not needed for this discussion - a constant for each nucleus). The big thing to glean from this equation is that the external field and the frequency are directly proportional. If the external field is larger , the frequency needed to induce the alpha to beta transition is larger. It follows then that in a larger field, higher frequency radio waves would be needed to induce the transition.
In this context, it is relevant to note that different nuclear magnetic resonance spectrometers have different magnetic field strengths. For example, the NMR on the first floor of Park Hall has a relatively high field, superconducting magnet. Because the field is high (high enough to erase bank cards and interfere with pacemakers and watches), the frequency range needed to excite protons is relatively high. It is called a 300 MHz (MHz = megahertz, a hertz is a cycle per second - a frequency unit) spectrometer, referring to the excitation frequency. The NMR on the second floor of Park Hall has a much weaker electromagnet associated with it. It is a 60 MHz instrument. Since different NMRs have different operating frequencies, spectra cannot be compared from different machines if they are reported in frequency units. For this reason, the universal ppm (parts per million) units are used in NMR. Please note the following relationship between ppm and frequency. The fact that frequency and ppm are directly proportional is all you need to retain for the future discussion and the course in general.
| Chemical shift in ppm = | peak position in Hz (relative to TMS) |
| spectrometer frequency in MHz |
Now let us use these basic ideas to better understand and interpret NMR spectra.
1. Why do we see peaks? When the excited nuclei in the beta orientation start to relax back down to the alpha orientation, a fluctuating magnetic field is created. This fluctuating field generates a current in a receiver coil that is around the sample. The current is electronically converted into a peak. It is the relaxation that actually gives the peak not the excitation.
2. Why do we see peaks at different positions? Realize that in principle, a peak will be observed for every magnetically distinct nucleus in a molecule. This happens because nuclei that are not in identical structural situations do not experience the external magnetic field to the same extent. The nuclei are shielded or deshielded due to small local fields generated by circulating sigma, pi and lone pair electrons.
To understand this concept better, consider a "run of the mill" hydrogen like that in ethane or methane. When this sort of hydrogen is placed in a magnetic field, the sigma electrons start to circulate. Remember : Magnetic fields generate currents. When the electrons circulate, they generate a small magnetic field that happens to point in the opposite direction to the external field. Remember: Currents have associated magnetic fields. Since magnetism is a vector quantity (vector quantities have direction and magnitude), this local field reduces the overall field somewhat. Therefore, the described hydrogen experiences a reduced magnetic field. If we reconsider the important NMR equation given on page two of this document, we can only conclude that if the external field is lower then the frequency of the electromagnetic radiation needed to induce the alpha to beta transition must be lower. Remember that frequency and ppm are directly proportional. Therefore, if a hydrogen requires a lower frequency, then it will show up as a peak at a lower ppm value. Hydrogens like those in methane are at around 1.0 ppm in the NMR spectrum.
|
|
| methane | bromoethane |
Now consider a hydrogen near a halogen as in bromoethane. This type of hydrogen is in a magnetically altered situation as compared to the hydrogen in methane. Due to its inherent electronegativity, the halogen atom has the effect of pulling sigma electron density away from the hydrogens in the molecule. The effect is largest for the hydrogens closest to the halogen atom. Though the little local opposing sigma field is still generated next to the hydrogens, it is partially pulled away by the electronegative bromine . Therefore, the hydrogens experience less of the local field and more of the external field. In other words, the vector in the vicinity of the hydrogen has been reduced as compared to methane. After you do the vector addition you end up with a larger overall field (again, as compared to methane). So going back to the fact that field and frequency are directly proportional, hydrogens near an electronegative atom should require a higher frequency to flip from the alpha to beta orientation. Therefore, they should appear at a higher ppm in the spectrum. Hydrogens like those in bromoethane should appear from ca. 2.5-4.0 ppm in the NMR spectrum.
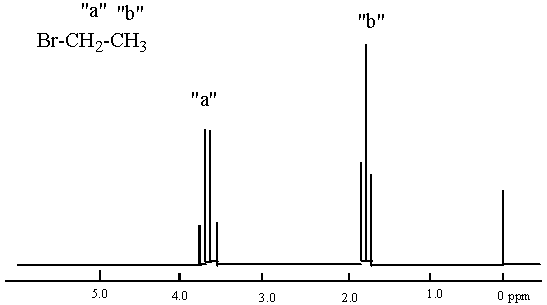
1
H NMR Spectrum of Bromoethane
Now as a last example, let us consider the NMR spectrum of benzene. Benzene and aromatics in general are very interesting because their hydrogens appear around 7 ppm even though they have no electronegative atoms. Why is this so? It has to do with the pi electrons. Because benzene and its relatives are aromatic, the p orbitals at each carbon in the ring overlap forming one continuous pi system. When the benzene ring is placed in a magnetic field, the external field induces a current in the pi system and that current generates a secondary magnetic field (or induced magnetic field). Once again, remember that electric currents have associated magnetic fields and that magnetic fields generate currents. The secondary magnetic field is such that it adds to the external field in the vicinity of the aromatic hydrogens as diagrammed below.
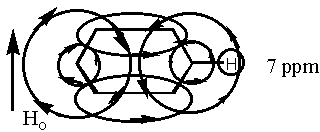
Benzene
If the local field is in the same direction as the external field, the resulting field is larger than the external field. This means that the frequency needed to flip those hydrogens experiencing that field is larger. Larger frequency translates into higher ppm position.
It is really interesting to consider 18-annulene diagrammed below. 18-Annulene is a large enough ring to have both cis and trans double bonds. This means that some of the hydrogens are pointing in toward the center of the aromatic ring. Reconsider the diagram of benzene above. If you look at it carefully, you will see that the magnetic field opposes the external field on the inside of the ring!!! If 18-annulene is aromatic like benzene, the inner hydrogens should absorb at lower frequency (ppm) and guess what? They do - they appear at -1.7 ppm!! Isn't that neat!!
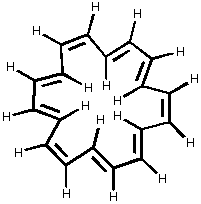
[18]-Annulene
So summing up, the different hydrogens of a molecule appear at different positions because small local magnetic fields are generated when local electrons begin to circulate due to the effect of the external magnetic field. These small fields either add to or subtract from the external field altering the frequency needed for excitation. Some of the effects are due to the circulation of sigma electrons while others are due to the circulation of pi electrons. The pi effects can be the most dramatic as was demonstrated in the preceding examples.
3. What causes splitting?
Many peaks in NMR spectra appear as symmetric patterns called doublets, triplets, quartets, quintets, etc. When you see these patterns it tells you about the number of adjacent (usually on the carbon next door to that bearing the absorbing hydrogen(s)), but different hydrogens. In simple spectra such as those we will be studying in organic chemistry lab, the number of peaks you see is one more than the number of adjacent, but different hydrogens. This is the so called n+1 rule. Different means that the adjacent hydrogens have a unique magnetic environment and absorb at a distinct frequency compared with the hydrogens in question. For example, consider bromoethane (structure given below).
Bromoethane has two different types of hydrogens so we expect two absorptions in the NMR spectrum. One absorption corresponds to the two hydrogens that are closest to the halogen atom. The other to the hydrogens comprising the methyl group that is farther away. Based on what I described above with regard to chemical shift (the ppm value), the hydrogens nearer the bromine should be at a higher ppm position. The hydrogens further from the bromine should be at lower ppm position.
Anyway, getting back the splitting, the hydrogens closer to the bromine will appear as a quartet because they are near three different hydrogens (the hydrogens on the methyl group). Those adjacent hydrogens are communicating their presence to the hydrogens being flipped. They are saying, "We are your neighbors and there are three of us." The reason they are able to communicate their presence is that they are little magnets and as such, they either add to or subtract from the external magnetic field depending on their orientation. Since there are many protons in a sample, the following are the possibilities for the neighboring hydrogens during excitation:
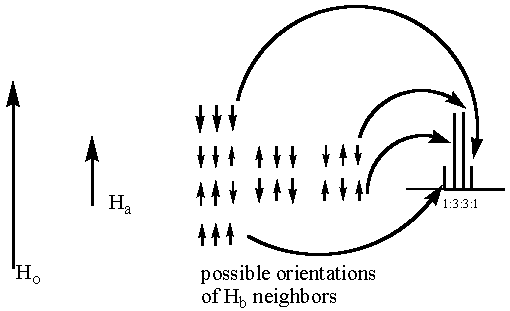
Please note that in the above diagram the "a" hydrogens are the ones near the bromine being flipped from the alpha to beta orientation. The "b" hydrogens are the three neighbors. As shown above, it is possible that a given "a" hydrogen will have three "b" hydrogens nearby that are aligned with the applied field during excitation. It is also possible that the three neighbors could all be aligned against the applied field. More probable is that either two protons will be aligned against the field or two with the field. These situations are more probable because there are more combinations of the three nuclei that give rise to these two possibilities. Since there are three combinations of each of these two, they are each three times more probable than having all three adjacent nuclei aligned with or against the field.
Now let us think about what these neighboring, local magnets do to the overall field. The "a" hydrogens that have all three neighbors aligned against the field have a lower overall magnetic field. Going back to the fundamental nuclear magnetic resonance equation (see page 2 ), you would conclude that these "a" hydrogens would have a lower frequency requirement for the alpha to beta transition and therefore appear at lower ppm. For the "a" hydrogens having three neighbors with all three "b" hydrogens aligned with the external field, the cumulative local field adds to the external field. This resultant field is larger than the external field so higher frequency electromagnetic radiation is needed to induce the alpha to beta transition. For the "b" hydrogens near two nuclei aligned with the field and one nucleus aligned against the field there is a slight increase in overall field leading to slightly higher frequency requirements. Similarly, two spins aligned against and one aligned with the field leads to slightly lower frequency requirements. So in the end, the "a" population is divided into four groups appearing at slightly different frequencies. The intermediate frequency peaks are taller than the higher and lower frequency peaks because they reflect more probable situations for local hydrogens. Hence, a quartet is observed.
Now if you understand why the "a" hydrogens give a quartet can you figure out why the "b" hydrogens give a triplet? Try to work it out using vectors as done in the above diagram.
For simple systems like bromoethane, n + 1 peaks will be observed for a given absorption, where n = the number of neighboring, but different hydrogens. This formula can be very useful when interpreting simple spectra.
The Interpretation of Simple NMR Spectra
This year, we will abstract the following information from NMR spectra to determine structures of products from organic reactions and isolations.
1. The number of peaks. The number of peaks is directly related to symmetry. If a compound has three significantly different types of hydrogens, it should have three different NMR absorptions.
2. The area under each absorption (the integral). The relative areas (or integrals) of the various absorptions in an NMR spectrum equals the relative number of hydrogens absorbing. If we know the molecular formula of a compound, we can use this ratio to figure out the actual number of each type of hydrogen. From the numbers of each type, we can infer the carbon structure. For example, with bromoethane, the relative areas under the NMR peaks are 2:3. This tells us that there is a group of two hydrogens that are the same and another group of three hydrogens that are the same.With your current knowledge of organic chemistry, it seems most likely that the compound has a methyl (-CH3) and a methylene (-CH2-) group. In other words, the most probable way to have three identical hydrogens is on a methyl group. The most probable way to have two identical hydrogens is in a methylene group.
Suppose you have a compound with the formula C5H12O and you are told that there are two NMR peaks, having the relative areas of 1:3. Can you come up with the structure of the compound?
3. The splitting pattern. For this semester, we will be using the n+1 rule as it applies to the simple structures we will be determining. You will see one more peak than the number of adjacent, but different hydrogens. Therefore, you can look at any peak and automatically know how many neighbors there are. This is crucial information because it allows you to start to hook atoms together in your structure. The problem is that people often confuse integral with splitting. So you must always remember this saying "Integral tells you what is here and splitting tells you what is near" This means that the integral tells you about the absorbing hydrogens and the splitting tells you about the neighbors. So what does it mean if you see a quintet with an area of two in a spectrum?
4. The position of the peak or the chemical shift ( ). This tells you about the electronic environment (the electronic environment directly relates to the magnetic environment) of the absorbing hydrogens. It will tell you if there are pi bonds or electronegative atoms nearby, etc. There are nice tables available that organize how different groups effect the frequency of absorptions and in lab you will always have these tables available to you. Yes, you will even have them on exams. A good rule of thumb when you are solving spectra is that the closer a hydrogen is to an electronegative atom the higher the ppm position. This little rule only works if the hydrogen is two or more bonds away from the atom. You will soon see the utility of this when you begin your problems in the workshop. It is also useful to keep in your head that aromatic hydrogens absorb at around 7 ppm.
A few tricks of the trade that are generally useful for spectral problem solving......
1. Always calculate the index of hydrogen deficiency or unsaturation number at the beginning of a problem ( you will normally be given the formula of the compound). Determining the unsaturation number is very helpful in regard to knowing which structural elements need to be present in your final solution. The unsaturation number is where you compare the actual formula with the theoretical saturated formula and compute the number of pairs of hydrogens that are missing. This topic should have been covered in class by now.
2. It is a good idea to interpret your IR spectrum before you do the NMR spectrum so that you have an idea about which functional groups are present in your molecule.
3. Organize your ideas about the structure of the unknown as you go along. For some people it is helpful to set up the following table for the NMR data and conclusions. The important part of the table is the conclusion column in which you are drawing a structural conclusion about the absorbing hydrogens and their neighbors. You should write a structural fragment down as has been done below for bromoethane.
| ppm | integral | splitting | conclusion |
| 1.6 | 3 | triplet | CH3CH2- |
| 3.4 | 2 | quartet | -CH2CH3 near electronegative atom |
4. You will notice as we do problems in class that we tend to emphasize and draw the most information from the integral and splitting. Chemical shift (ppm position) in many cases is the last point of interest. There are a few relevant chemical shifts that should be interpreted at the outset of a problem.. One is the aromatic chemical shift. Aromatic hydrogens absorb at ca. 7 ppm. This is a very distinct and characteristic shift and should be interpreted immediately. If you observe a peak at seven chances are you have an aromatic ring. The most common aromatic ring is benzene. Another very distinctive shift is that of the aldehyde functional group. Aldehydic hydrogens appear at ca. 9 ppm in the spectrum. if you see a shift of nine ppm assume that you have an aldehyde functional group.
5. Solving spectra rapidly involves making good educated guesses. If you get an integral of three there is really only one probable way to have three identical hydrogens - a methyl group. If you get an integral of nine it is most likely three methyl groups that are the same by symmetry. If you get aromatic absorptions, you probably have one or more benzene rings. Always start with the simplest ideas and work your way toward more exotic solutions.
If you want to discuss any of this please feel free to stop by.
[Back to Study Aids]